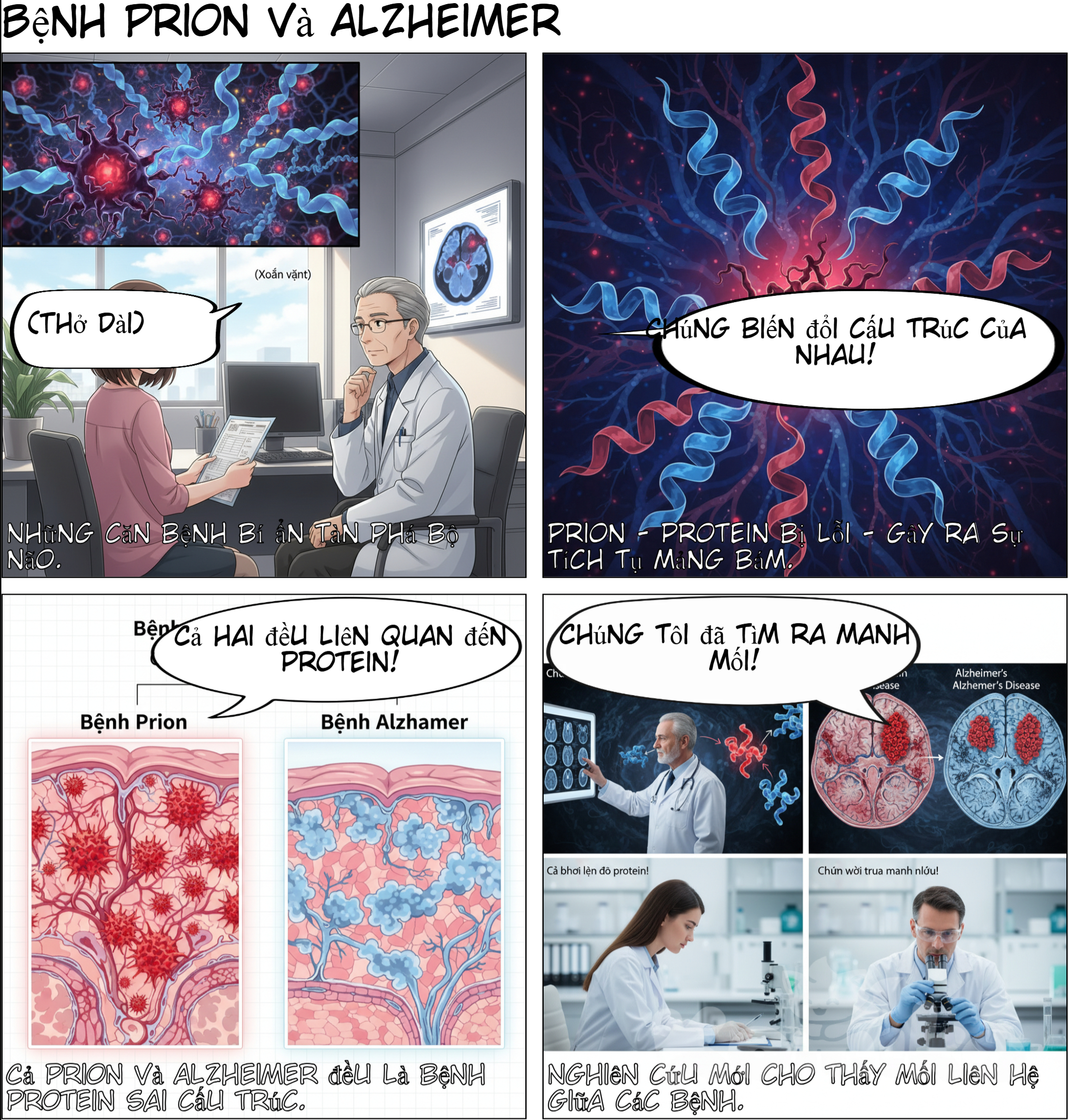
AI 아트: Prion Diseases and Characteristics Prion diseases are part of a class of illnesses known as transmissible spongiform encephalopathies (TSEs). Symptoms—including loss of coordination, behavioral changes, and dementia—are strikingly similar to Alzheimer's. A key difference is that prion diseases are usually rapidly fatal, often ending in death within 12 months after symptoms first develop. Misfolding Protein Mechanism Prion diseases develop due to malign structural features in proteins. A prion is a misfolded protein. This misfolded protein can be introduced exogenously (as an infectious agent) or endogenously (via an inherited PRNP gene). When the misfolded prion enters the central nervous system, it adheres to other proteins and causes them to become misshapen. This process results in plaque buildup in the brain, which blocks the normal passage of nerve signals. Connection to Alzheimer's Disease (AD) Both prion diseases and Alzheimer’s disease are classified as “misfolding protein diseases”. In AD, the characteristic plaques are caused by amyloid-ß peptides. In all these misfolding diseases (including Parkinson's and Huntington’s), the protein accumulation begins with a single “bad seed” ready to propagate its problematic form. Evidence of Amyloid-ß Transmission In 2015 and 2016 studies, researchers examined cadavers of patients who had acquired Creutzfeldt-Jakob disease (CJD)—a prion disease—through iatrogenic means (in the course of medical treatment), such as surgical grafts of dura mater or contaminated growth hormones. The brain tissue of these CJD patients showed the expected prion damage but also revealed initial signs of amyloid-ß plaques. This was unusual because none of the patients’ medical history indicated they had developed Alzheimer’s disease. The studies suggested evidence that amyloid-ß was also transmitted through the same medical procedures that gave them CJD hãy tạo truyện tranh cho tôi dựa trên nội dung này
생성자 bouncy marshmallow
콘텐츠 세부 정보
미디어 정보
사용자 상호작용
이 AI 작품에 대하여
설명
창작 프롬프트
참여

bouncy marshmallow
bouncy marshmallow
Prion Diseases and Characteristics Prion diseases are part of a class of illnesses known as transmissible spongiform encephalopathies (TSEs). Symptoms—including loss of coordination, behavioral changes, and dementia—are strikingly similar to Alzheimer's. A key difference is that prion diseases are usually rapidly fatal, often ending in death within 12 months after symptoms first develop. Misfolding Protein Mechanism Prion diseases develop due to malign structural features in proteins. A prion is a misfolded protein. This misfolded protein can be introduced exogenously (as an infectious agent) or endogenously (via an inherited PRNP gene). When the misfolded prion enters the central nervous system, it adheres to other proteins and causes them to become misshapen. This process results in plaque buildup in the brain, which blocks the normal passage of nerve signals. Connection to Alzheimer's Disease (AD) Both prion diseases and Alzheimer’s disease are classified as “misfolding protein diseases”. In AD, the characteristic plaques are caused by amyloid-ß peptides. In all these misfolding diseases (including Parkinson's and Huntington’s), the protein accumulation begins with a single “bad seed” ready to propagate its problematic form. Evidence of Amyloid-ß Transmission In 2015 and 2016 studies, researchers examined cadavers of patients who had acquired Creutzfeldt-Jakob disease (CJD)—a prion disease—through iatrogenic means (in the course of medical treatment), such as surgical grafts of dura mater or contaminated growth hormones. The brain tissue of these CJD patients showed the expected prion damage but also revealed initial signs of amyloid-ß plaques. This was unusual because none of the patients’ medical history indicated they had developed Alzheimer’s disease. The studies suggested evidence that amyloid-ß was also transmitted through the same medical procedures that gave them CJD hãy tạo truyện tranh cho tôi dựa trên nội dung này
4 months ago